Answer of June 2019
For completion of the online quiz, please visit the HKAM iCMECPD website: http://www.icmecpd.hk/
Clinical History:
21-year-old girl complained of headache and gradual hearing loss.
MRI of the brain and spine were performed.
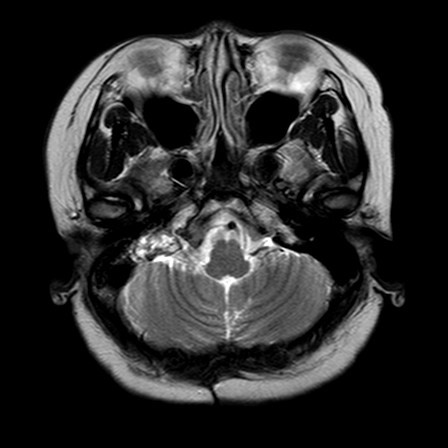
Image 1 (T2W axial)
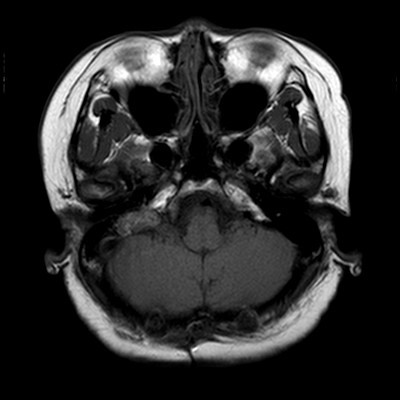
Image 2 (T1W axial)
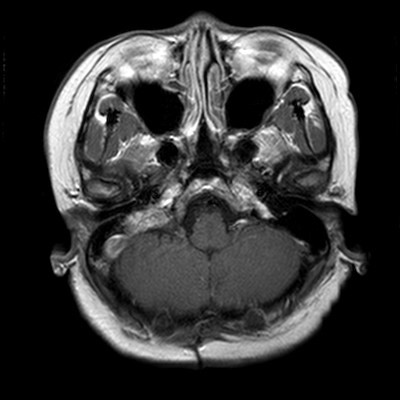
Image 3 (T1W with contrast axial)
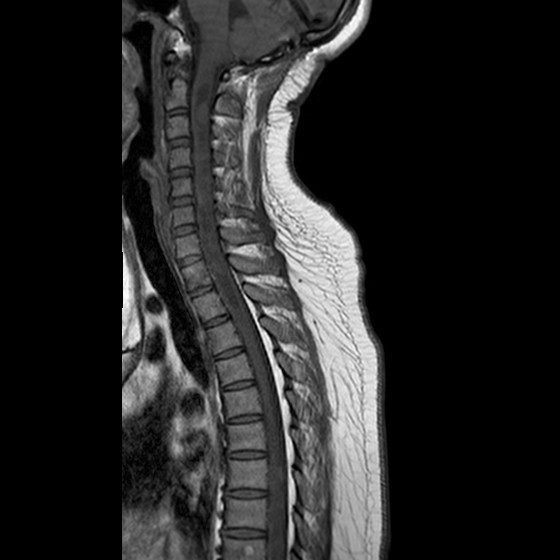
Image 4 (T1W sagittal)
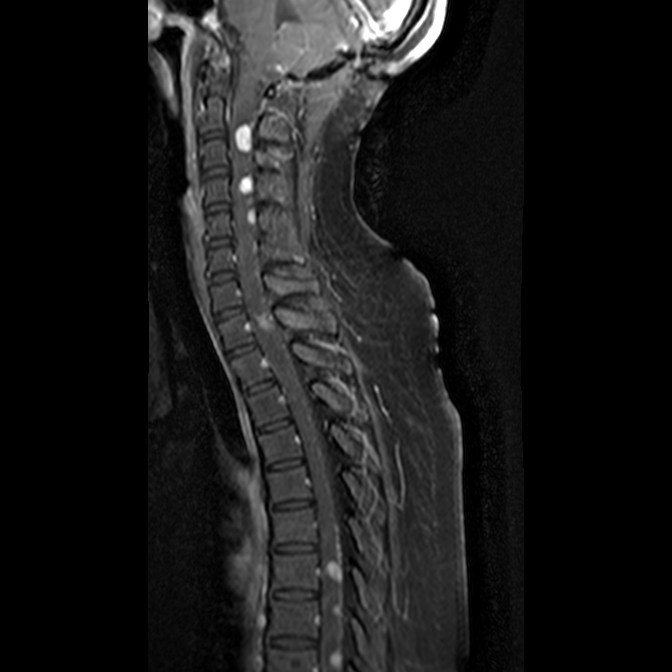
Image 5 (T1W with contrast sagittal)
Imaging Findings:
- Avidly enhancing heterogeneous T1 and T2 hyperintense lesion centered at the right petrous temporal bone protruding into the right cerebellopontine angle with adjacent involvement of the right internal auditory canal and semicircular canals. Mild mass effect on the ipsilateral cerebellar peduncle.
- Retention changes in right mastoid air cells.
- Supratentorial brain parenchyma unremarkable (not shown).
- Multiple T1 isointense T2 hyperintense intramedullary nodular lesions along the spinal cord. Avid enhancement detected.
Diagnosis:
Von Hippel-Lindau syndrome with MR features of right endolymphatic sac tumor and multiple spinal hemangioblastomas.
Discussion:
Von Hippel-Lindau syndrome (VHL) is a hereditary condition with an estimated prevalence of 1 in 36 000. It is characterized by a germline mutation in chromosome 3p25 – 26, which involves the VHL tumor suppressor gene. The inheritance is autosomal dominant accounting for 80% of cases, while 20% of cases arise from de novo mutations.
Patients develop multi-organ benign and malignant tumors involving the central nervous system (CNS), head, pancreas, kidneys, adrenals, and reproductive organs. In the CNS, hemangioblastomas can involve the retina, cerebellum, or spinal cord. Endolymphatic sac tumors are seen affecting the temporal bone. Cysts, serous cystadenomas, and neuroendocrine tumors can involve the pancreas. In the kidneys, multiple renal cysts and renal cell carcinomas (RCC) of the clear cell type can occur. Adrenal pheochromocytomas and extra-adrenal pheochromocytomas/paragangliomas may arise in VHL patients. In the reproductive organs, epididymal papillary cystadenomas can develop in men, while papillary cystadenomas can manifest in women.
Imaging plays a key role in the diagnosis and surveillance of the condition. For CNS hemangioblastomas, lesions typically are cystic with an avidly enhancing mural nodule seen in CT and MR imaging. Flow voids may be detected on MRI. Endolymphatic sac tumors appear as moth-eaten lytic lesions involving the petrous temporal bone with local erosive changes of adjacent structure in CT scan, while manifesting as T1 and T2 hyperintense lesion (secondary to hemorrhagic component) with avid enhancement. Pancreatic serous cystadenomas classically appear as multilocular small cystic lesions with thin fibrous septa and a central scar showing stellate calcification. Pancreatic neuroendocrine tumors are typically solid and demonstrates avid early arterial enhancement with washout on delayed phase in CT and MR images. Clear cell RCCs appear as heterogeneous enhancing masses. They may demonstrate signal loss with out-of-phase MR sequence due to presence of intracellular lipid. Pheochromocytomas are typically solid lesions showing avid arterial enhancement in CT images and showing marked T2 hyperintensity on MRI (“light bulb” sign). MIBG scintigraphy can be performed for detection of pheochromocytomas, which demonstrate avid tracer uptake.
Diagnosis can be made clinically when clinical history and findings manifest or by genetic testing.
Treatment of VHL syndrome is complex, which requires expertise from various specialties to address the multi-organ involvement. Early identification of lesions is important for facilitating optimal management in order to reduce morbidity and mortality. Genetic testing and counselling for diagnosis and for at-risk pregnancies should be made available. Surveillance should be conducted for patients. Various surveillance guidelines exist. One such guideline is the VHL Alliance Surveillance Guideline which suggests surveillance start at birth and continue lifelong.